Wilson Disease
Summary
Wilson disease is an autosomal recessive disorder of copper metabolism caused by mutations in the ATP7B gene, leading to copper accumulation in the liver, brain, cornea, and other organs. It typically presents in childhood to early adulthood with hepatic disease (ranging from asymptomatic liver enzyme elevation to cirrhosis or fulminant hepatic failure), neuropsychiatric symptoms (tremor, dystonia, parkinsonism, psychiatric disturbances), or both. The pathognomonic finding is Kayser-Fleischer rings (copper deposits in Descemet's membrane). Diagnosis is supported by low serum ceruloplasmin, elevated 24-hour urinary copper, and genetic testing. Treatment with copper chelation (D-penicillamine, trientine) or zinc is lifelong and can prevent progression. Liver transplantation is curative for hepatic Wilson disease.
Key Facts
- Definition: Autosomal recessive copper accumulation disorder (ATP7B gene)
- Incidence: 1 in 30,000 live births; carrier frequency 1:90
- Peak Demographics: Presentation 3-40 years (most 5-35)
- Pathognomonic: Kayser-Fleischer rings + low ceruloplasmin
- Gold Standard Investigation: Leipzig Score (ceruloplasmin, urinary copper, KF rings, liver copper, genetics)
- First-line Treatment: D-penicillamine or trientine (chelation) + zinc
- Prognosis: Excellent if treated early; fatal if untreated
Clinical Pearls
Diagnostic Pearl: Kayser-Fleischer rings are seen in >95% of neurological Wilson but only 50% of hepatic Wilson - absence does not exclude.
Emergency Pearl: Fulminant Wilson disease = acute liver failure + Coombs-negative haemolysis. Emergency transplant required.
Treatment Pearl: Neurological symptoms may initially worsen with chelation therapy - warn patients.
Why This Matters Clinically
Wilson disease is treatable and fatal if untreated. It should be considered in any young patient with liver disease, movement disorder, or psychiatric symptoms of unclear cause.
Incidence
- 1 in 30,000 live births
- Carrier frequency: 1 in 90
- Equal sex distribution
Age of Presentation
- Hepatic: Usually 5-15 years
- Neurological: Usually 15-30 years
- Range: 3-40 years (rarely older)
Mechanism
Step 1: Genetic Defect
- Mutations in ATP7B gene (chromosome 13)
- ATP7B is a copper-transporting ATPase in hepatocytes
Step 2: Impaired Copper Excretion
- Copper cannot be incorporated into ceruloplasmin
- Copper cannot be excreted into bile
- Low serum ceruloplasmin
Step 3: Copper Accumulation
- Copper accumulates in liver (first)
- Then releases into blood, deposits in brain, cornea, kidneys
Step 4: Organ Damage
- Liver: Oxidative damage, inflammation, fibrosis, cirrhosis
- Brain: Basal ganglia damage (lenticular nuclei)
- Cornea: Kayser-Fleischer rings
- RBCs: Haemolysis
Hepatic
Neurological
Psychiatric
Other
Red Flags
[!CAUTION]
- Young patient with cirrhosis
- Acute liver failure with haemolysis
- Neuropsychiatric symptoms + liver disease
- Coombs-negative haemolysis
Screening Tests
| Test | Finding | Notes |
|---|---|---|
| Ceruloplasmin | Low (less than 0.2 g/L) | 90% have low levels |
| 24h urinary copper | Elevated (greater than 100 mcg/day) | Greater than 40 mcg is suggestive |
| Slit-lamp exam | Kayser-Fleischer rings | 95% neurological, 50% hepatic |
Leipzig Score
Points from:
- KF rings (present = 2)
- Neurological symptoms (= 2)
- Coombs-negative haemolysis (= 1)
- Urinary copper (= 0-2)
- Liver copper (= 0-2)
- Ceruloplasmin (= 0-2)
- ATP7B mutations (= 4 if 2 mutations)
Score 4 or more = Wilson disease
Imaging
- MRI Brain: Basal ganglia T2 hyperintensity, "face of the giant panda" sign
- Liver imaging: Hepatomegaly, cirrhosis
Liver Biopsy
- Hepatic copper content greater than 250 mcg/g dry weight
- Histology: Varying stages of liver disease
Genetic Testing
- ATP7B gene sequencing
- Confirms diagnosis if 2 pathogenic mutations
Algorithm
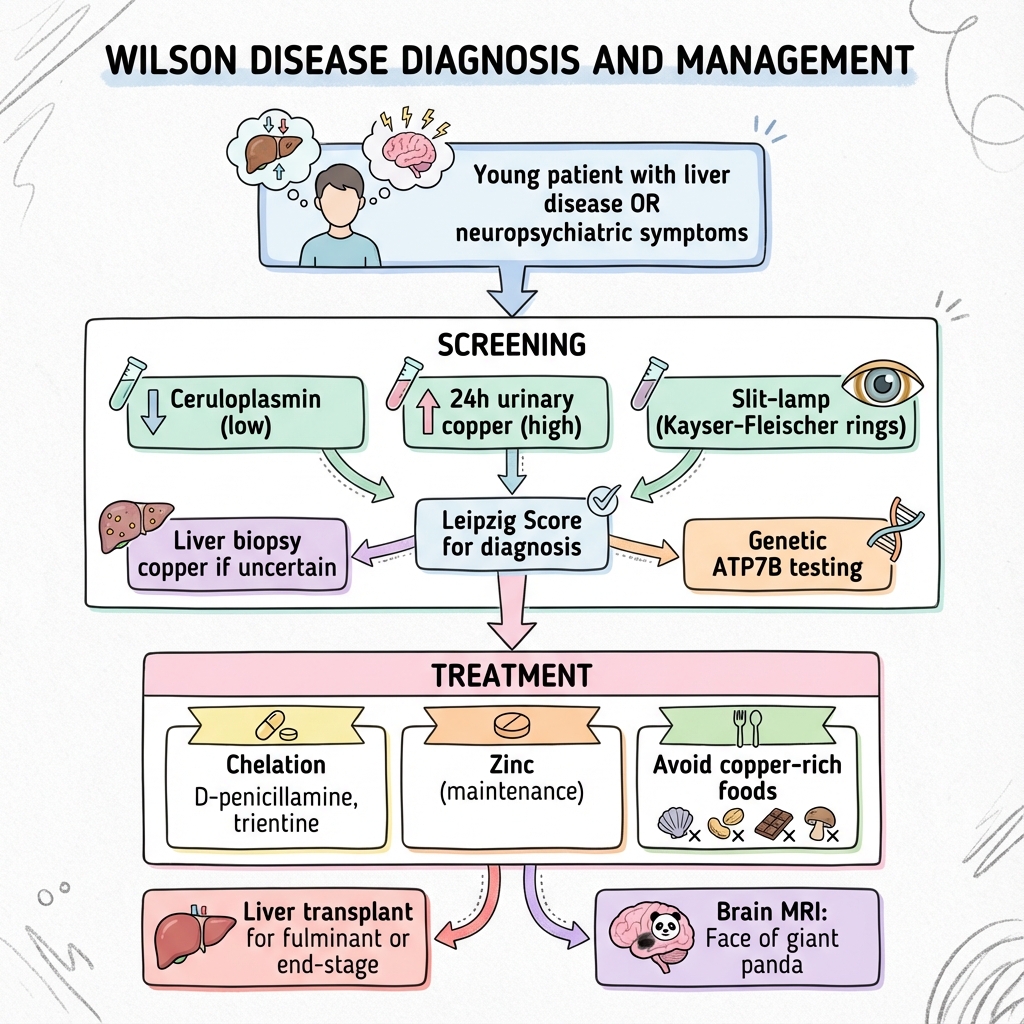
Chelation Therapy
| Drug | Dose | Notes |
|---|---|---|
| D-penicillamine | 250-500mg QDS | First-line; monitor for side effects |
| Trientine | 250-500mg TDS | Alternative if penicillamine intolerant |
Zinc Therapy
| Drug | Dose | Notes |
|---|---|---|
| Zinc acetate | 50mg TDS | Maintenance; blocks copper absorption |
Approach
- Initial: Chelation therapy (D-penicillamine or trientine)
- Maintenance: Zinc +/- lower dose chelation
- Lifelong treatment required
- Monitor urinary copper, LFTs, FBC
Liver Transplantation
Indications:
- Fulminant hepatic failure
- Decompensated cirrhosis not responding to treatment
- Curative (corrects metabolic defect)
Dietary Advice
- Avoid high-copper foods: Shellfish, liver, chocolate, nuts, mushrooms
- Avoid copper utensils
Monitoring
- 24h urinary copper (aim for normal on treatment)
- LFTs, ceruloplasmin
- FBC (penicillamine side effects)
- Neurological assessment
Outcomes
- With early treatment: Normal life expectancy
- Without treatment: Fatal (liver failure or neurological progression)
- Neurological recovery: Variable; may take years
- Hepatic: Good response if pre-cirrhotic
-
Roberts EA et al. AASLD Practice Guidelines: Wilson Disease. Hepatology. 2008;47(6):2089-2111. PMID: 18506894
-
European Association for Study of the Liver. EASL Clinical Practice Guidelines: Wilson's Disease. J Hepatol. 2012;56(3):671-685. PMID: 22340672
-
Weiss KH, Stremmel W. Clinical considerations for an effective medical therapy in Wilson's disease. Ann N Y Acad Sci. 2014;1315:81-85. PMID: 24820352
Viva Points
"Wilson disease is autosomal recessive copper accumulation (ATP7B). Presents with hepatic/neuropsychiatric disease in young adults. Diagnose with low ceruloplasmin, high urinary copper, KF rings. Treat with chelation (D-penicillamine, trientine) + zinc. Transplant for fulminant/end-stage."
Key Facts
- ATP7B gene mutation
- KF rings: 95% neurological, 50% hepatic
- Fulminant Wilson = liver failure + Coombs-negative haemolysis
- Face of giant panda sign on MRI
- Treatment is lifelong
Common Mistakes
- Missing diagnosis in young cirrhosis
- Not screening siblings
- Stopping treatment (relapse)
Last Reviewed: 2026-01-01 | MedVellum Editorial Team