Cardiac Amyloidosis
Summary
Cardiac amyloidosis is an infiltrative cardiomyopathy caused by extracellular deposition of misfolded proteins (amyloid fibrils) in the myocardium. Previously considered rare, improved recognition and diagnostic techniques have revealed it to be an under-diagnosed cause of heart failure with preserved ejection fraction (HFpEF), particularly in elderly patients. The two main types are light-chain (AL) amyloidosis and transthyretin (ATTR) amyloidosis, with ATTR further subdivided into hereditary (ATTRv, variant) and wild-type (ATTRwt, formerly senile cardiac amyloidosis). Early diagnosis is critical as prognosis differs markedly between types and disease-modifying therapies (tafamidis) are now available for ATTR. The classic presentation of low-voltage ECG despite increased wall thickness on echocardiography should prompt investigation.
Key Facts
- Definition: Infiltrative cardiomyopathy from extracellular amyloid fibril deposition causing diastolic dysfunction and restrictive physiology
- Incidence: AL amyloidosis 10-12 per million/year; ATTRwt found in 13-25% of HFpEF patients >60 years
- Mortality: AL cardiac amyloidosis median survival 6 months untreated; ATTR 2-5 years
- Peak Demographics: AL 50-70 years; ATTRwt predominantly males >65 years; ATTRv variable by mutation
- Pathognomonic Feature: Low-voltage ECG with increased LV wall thickness on echo
- Gold Standard Investigation: Cardiac MRI with late gadolinium enhancement + bone scintigraphy (Tc-99m PYP/DPD)
- First-line Treatment: AL - chemotherapy; ATTR - tafamidis (disease-modifying)
- Prognosis: Improved significantly with early diagnosis and targeted therapy
Clinical Pearls
Diagnostic Pearl: The combination of LVH on echo with low-voltage QRS on ECG is highly suggestive of infiltrative disease. Request cardiac MRI and bone scintigraphy.
Examination Pearl: Bilateral carpal tunnel syndrome preceding heart failure symptoms by years is characteristic of ATTR amyloidosis due to amyloid deposition in flexor retinaculum.
Treatment Pearl: Tafamidis reduces mortality and hospitalisation in ATTR cardiac amyloidosis by 30% - ensure all patients are evaluated for this therapy.
Pitfall Warning: Do not assume all amyloidosis is AL. ATTR is more common than previously thought and has different treatment (no chemotherapy needed).
Mnemonic: ATTR - Aged (wild-type), Transthyretin, Tafamidis treats, Runs in families (variant)
Why This Matters Clinically
Cardiac amyloidosis is increasingly recognised as a treatable cause of HFpEF. Missed diagnosis leads to inappropriate management and poor outcomes. With tafamidis now licensed for ATTR, early identification fundamentally changes prognosis. This is an emerging MRCP topic reflecting advances in cardiac imaging and therapeutics.
Incidence and Prevalence
- AL amyloidosis: 10-12 per million per year; cardiac involvement in 50-70%
- ATTRwt: Found in 13-25% of HFpEF patients >60 years; 16% of TAVR patients
- ATTRv: Prevalence varies by mutation; V122I present in 3-4% of African Americans
- Trend: Increasing recognition due to improved awareness and non-invasive diagnostics
- Geographic: ATTRv endemic in certain populations (Portugal, Sweden, Japan)
Demographics
| Factor | Details | Clinical Significance |
|---|---|---|
| Age | AL: 50-70; ATTRwt: >65 | ATTRwt almost exclusively elderly |
| Sex | ATTRwt male predominant 80% | AL more equal |
| Ethnicity | V122I in 3-4% African Americans | Screen high-risk populations |
| Geography | ATTRv endemic areas known | Family screening important |
| Occupation | None specific | - |
Risk Factors
| Factor | Type | Mechanism |
|---|---|---|
| Plasma cell dyscrasia | AL | Monoclonal light chains |
| TTR gene mutations | ATTRv | Unstable transthyretin |
| Advanced age | ATTRwt | Age-related TTR instability |
| Male sex | ATTRwt | Unknown |
| African American ancestry | ATTRv | V122I mutation prevalence |
| MGUS | AL | Precursor to myeloma |
Mechanism
Step 1: Amyloid Precursor Production
- AL: Clonal plasma cells produce excess monoclonal light chains (usually lambda)
- ATTR: Transthyretin (produced by liver) misfolds due to genetic mutation (ATTRv) or aging (ATTRwt)
- TTR normally transports thyroxine and retinol-binding protein
Step 2: Protein Misfolding and Fibril Formation
- Amyloidogenic proteins misfold into beta-pleated sheet configuration
- Oligomers aggregate into insoluble fibrils
- Fibrils are 7.5-10nm diameter, rigid, unbranched
- Characteristic apple-green birefringence under polarised light with Congo red staining
Step 3: Cardiac Infiltration
- Amyloid deposits in myocardial interstitium
- Progressive accumulation causes wall thickening without true hypertrophy
- Deposition also in coronary arteries, conduction system, atria
- Myocyte architecture disrupted, contractile function impaired
Step 4: Diastolic Dysfunction and Restrictive Physiology
- Stiff, non-compliant ventricles
- Elevated filling pressures
- Preserved or mildly reduced EF initially
- Atrial dysfunction and AF common
- Small cavity size despite thick walls
Step 5: Progressive Heart Failure
- Restrictive cardiomyopathy phenotype
- Right heart failure often predominant
- Conduction disease (AV block, AF)
- Intracardiac thrombus despite sinus rhythm
- Without treatment: progressive decline and death
Classification
By Amyloid Type:
| Type | Precursor | Features | Prognosis |
|---|---|---|---|
| AL (primary) | Immunoglobulin light chain | Multi-organ, rapid; nephrotic syndrome | Median 6mo untreated |
| ATTRwt (wild-type) | Wild-type transthyretin | Elderly males, carpal tunnel | Median 3-5 years |
| ATTRv (hereditary) | Mutant transthyretin | Family history, neuropathy | Variable by mutation |
| AA (secondary) | Serum amyloid A | Chronic inflammation; rare cardiac | Treat underlying cause |
By Cardiac Stage (Mayo Staging for AL):
| Stage | Criteria | Median Survival |
|---|---|---|
| I | TnT less than 0.025, NT-proBNP less than 332 | 26 months |
| II | Either elevated | 11 months |
| III | Both elevated | 4 months |
Symptoms
Typical:
Extracardiac Clues:
Signs
Red Flags
[!CAUTION]
- Low-voltage ECG + LVH on echo
- HFpEF with bilateral carpal tunnel history
- Unexplained LVH in elderly
- Nephrotic syndrome + heart failure
- Syncope in HFpEF patient
Structured Approach
General:
- Cachexia (AL)
- Periorbital purpura (AL)
- Macroglossia (AL - protrude tongue)
- Carpal tunnel scars
Cardiovascular:
- JVP: elevated, Kussmaul's sign (rises on inspiration)
- Apex: non-displaced, difficult to palpate
- Auscultation: soft S1/S2, S4, no significant murmur
- Signs of right heart failure
Peripheral:
- Peripheral oedema
- Hepatomegaly
- Orthostatic hypotension (check lying/standing BP)
Special Tests
| Test | Finding | Significance |
|---|---|---|
| Standing BP | >20mmHg systolic drop | Autonomic involvement |
| Carpal tunnel examination | Positive Tinel's/Phalen's | Early ATTR sign |
| Tongue examination | Macroglossia, teeth marks | AL amyloidosis |
| Skin examination | Periorbital purpura | AL amyloidosis |
First-Line
- ECG: Low voltage (limb leads less than 5mm), pseudo-infarct pattern, AF
- Urinalysis: Proteinuria (AL with renal involvement)
Laboratory
| Test | Finding | Purpose |
|---|---|---|
| NT-proBNP | Markedly elevated | Prognosis, monitoring |
| Troponin | Often elevated | Prognosis (Mayo staging) |
| eGFR | May be reduced | Renal involvement |
| LFTs | May show elevated ALP | Hepatic involvement |
| Serum free light chains | Abnormal ratio in AL | Distinguish AL from ATTR |
| Serum/urine immunofixation | Monoclonal protein in AL | Detect plasma cell dyscrasia |
| Genetic testing | TTR mutations | Confirm ATTRv |
Imaging
| Modality | Findings | Indication |
|---|---|---|
| Echocardiography | Increased wall thickness, granular/speckled myocardium, diastolic dysfunction, biatrial enlargement | First-line |
| Cardiac MRI | Late gadolinium enhancement (subendocardial, diffuse), abnormal T1 mapping, elevated ECV | Tissue characterisation |
| Bone scintigraphy | Tc-99m PYP/DPD/HMDP uptake (Grade 2-3) | Diagnose ATTR without biopsy |
| PET-CT | May show cardiac uptake | Emerging |
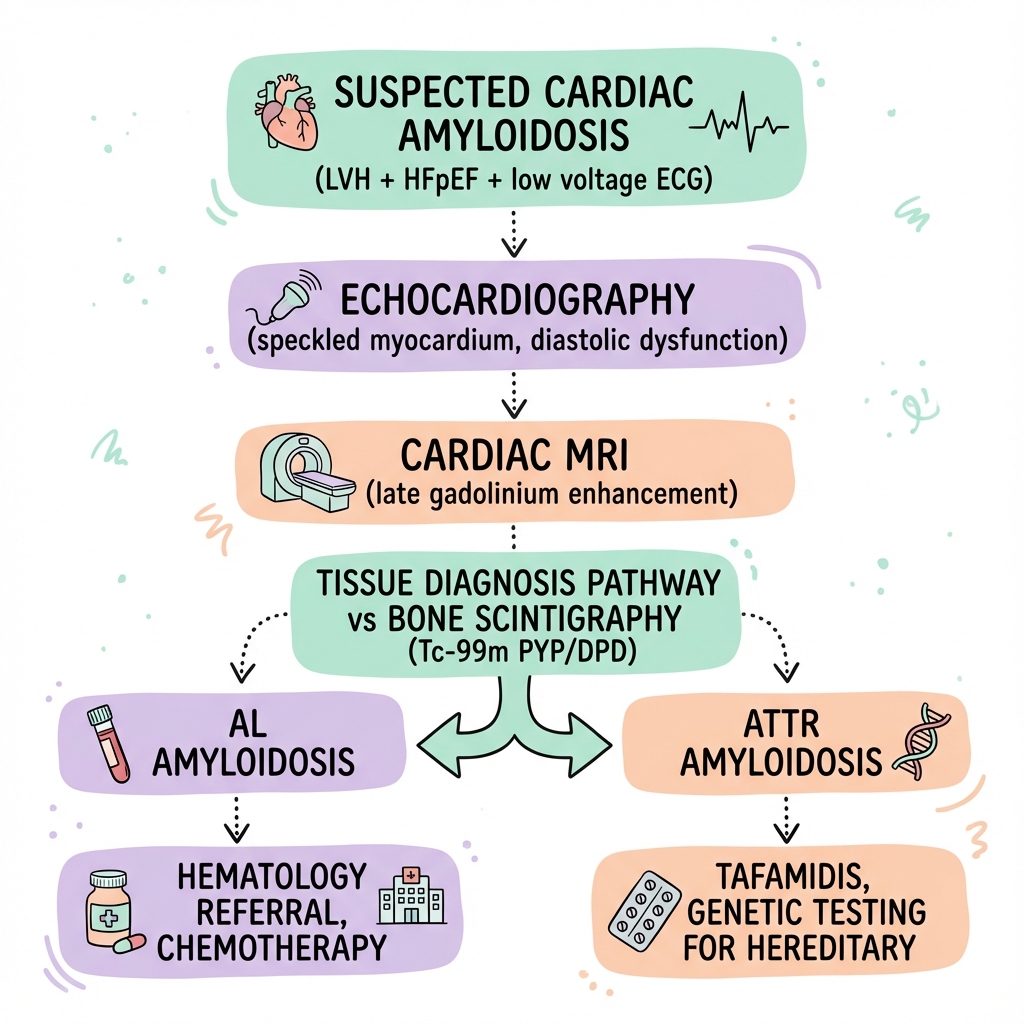
Diagnostic Algorithm
- Clinical suspicion: HFpEF + LVH + low voltage ECG
- Echocardiography: wall thickness, diastolic function
- Serum/urine immunofixation + free light chains
- If negative for AL → Bone scintigraphy
- Grade 2-3 uptake + no monoclonal protein = ATTR (no biopsy needed)
- If scintigraphy negative or monoclonal protein present → Tissue biopsy (endomyocardial or fat pad)
- If ATTR confirmed → Genetic testing for ATTRv
Management Algorithm
Conservative Management
- Fluid restriction 1.5-2L/day
- Low sodium diet
- Compression stockings for oedema
- Avoid dehydration (preload dependent)
- Fall prevention (autonomic dysfunction)
Medical Management
Heart Failure Therapy:
| Drug | Recommendation | Caution |
|---|---|---|
| Diuretics | Mainstay for congestion | Avoid over-diuresis |
| Loop diuretics | Furosemide/Bumetanide | Careful titration |
| MRAs | Spironolactone 25mg | Monitor K+ |
| Beta-blockers | Often poorly tolerated | Avoid if rate-dependent CO |
| ACEi/ARBs | Often poorly tolerated | Postural hypotension |
| Digoxin | AVOID or use very low dose | Binds amyloid fibrils, toxicity |
| CCBs | AVOID (negative inotropy) | Bind amyloid, toxicity |
Disease-Modifying Therapy:
| Type | Treatment | Evidence |
|---|---|---|
| ATTR | Tafamidis 80mg/61mg daily | ATTR-ACT trial: 30% mortality reduction |
| ATTR | Patisiran, Inotersen (with neuropathy) | RNA interference |
| AL | Chemotherapy (VCd, daratumumab) | Haematology-led |
| AL | Autologous stem cell transplant | Selected patients |
Surgical Management
- Heart transplant: Selected patients, especially younger ATTRv
- Combined heart-liver transplant: ATTRv (removes source)
- LVAD: Generally not recommended (restrictive physiology)
- Pacemaker: For conduction disease
Disposition
- Admit: New diagnosis, decompensated HF, syncope
- Discharge: Stable on therapy, outpatient follow-up arranged
- Follow-up: Cardiology 4-6 weekly initially; haematology for AL
Immediate
| Complication | Incidence | Management |
|---|---|---|
| Acute decompensated HF | Common | IV diuretics |
| AF with rapid response | 40-60% | Rate control (difficult) |
| Syncope | 20% | Evaluate for arrhythmia |
Early
- Intracardiac thrombus (even in sinus rhythm)
- AV block requiring pacing
- Ventricular arrhythmias
Late
- End-stage heart failure
- Multi-organ involvement (AL)
- Sudden cardiac death
- Progressive renal failure (AL)
Natural History
Without treatment, cardiac amyloidosis is progressive and fatal. AL cardiac amyloidosis has median survival of 6 months untreated. ATTRwt has slower progression with median survival 3-5 years.
Outcomes with Treatment
| Type | Treatment | Outcome |
|---|---|---|
| AL | Haematological response | Median survival 3-5 years |
| ATTR | Tafamidis | 30% reduction mortality |
| ATTR | No therapy | Median 3-5 years |
Prognostic Factors
Good:
- Early diagnosis
- ATTR (vs AL)
- Haematological response in AL
- Tafamidis-treated ATTR
Poor:
- Advanced cardiac stage (Mayo III)
- AL with no haematological response
- NYHA IV
- Syncope
Key Guidelines
- ESC Guidelines on Cardiomyopathies (2023) — Integrated approach to diagnosis and management
- AHA Scientific Statement on Cardiac Amyloidosis (2021) — Comprehensive review PMID: 34233884
- UK National Amyloidosis Centre Protocols — Expert centre guidance
Landmark Trials
ATTR-ACT Trial (2018) — Tafamidis for ATTR
- n=441 ATTR cardiac amyloidosis
- Tafamidis vs placebo
- Finding: 30% reduction all-cause mortality, 32% reduction CV hospitalisations
- Impact: First disease-modifying therapy for cardiac amyloidosis
- PMID: 30145931
APOLLO Trial (2018) — Patisiran for ATTRv
- n=225 hereditary ATTR with polyneuropathy
- Patisiran (siRNA) vs placebo
- Finding: Improved neuropathy and cardiac parameters
- PMID: 29972753
What is Cardiac Amyloidosis?
Cardiac amyloidosis is a condition where abnormal proteins build up in your heart muscle. These proteins are called amyloid. They make your heart stiff and unable to relax properly, which means it can't fill with blood effectively.
Why does it matter?
Without treatment, amyloid build-up gets worse over time and leads to heart failure. However, we now have treatments that can slow or stop the disease, especially if caught early.
How is it treated?
- Diuretics (water tablets) to remove excess fluid
- Tafamidis - a medication that stabilises the abnormal protein (for ATTR type)
- Chemotherapy - for AL type to stop the abnormal protein being made
- Careful monitoring with regular scans and blood tests
When to seek help
- Worsening breathlessness
- Increasing swelling
- Dizziness or fainting
- Palpitations
-
Maurer MS et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy (ATTR-ACT). N Engl J Med. 2018;379(11):1007-1016. PMID: 30145931
-
Kittleson MM et al. Cardiac Amyloidosis: An AHA Scientific Statement. Circulation. 2021;144(12):e185-e212. PMID: 34233884
-
Adams D et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis (APOLLO). N Engl J Med. 2018;379(1):11-21. PMID: 29972753
-
Gillmore JD et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016;133(24):2404-2412. PMID: 27143678
-
Garcia-Pavia P et al. Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC Working Group. Eur Heart J. 2021;42(16):1554-1568. PMID: 33825853
-
Dispenzieri A et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22(18):3751-3757. PMID: 15365071
-
González-López E et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-2594. PMID: 26224076
-
Fontana M et al. Native T1 Mapping in Transthyretin Amyloidosis. JACC Cardiovasc Imaging. 2014;7(2):157-165. PMID: 24412190
Common Exam Questions
- "72-year-old man with HFpEF and bilateral carpal tunnel. What is the likely diagnosis?"
- "How do you differentiate AL from ATTR amyloidosis?"
- "What is the significance of bone scintigraphy in cardiac amyloidosis?"
- "What medications should be avoided in cardiac amyloidosis?"
Viva Points
Opening:
"Cardiac amyloidosis is an infiltrative cardiomyopathy caused by amyloid fibril deposition. The two main types are AL (light chain) and ATTR (transthyretin). ATTR is more common than previously thought, found in 13-25% of HFpEF patients over 60."
Key Facts:
- Low-voltage ECG + LVH = red flag for infiltration
- Bone scintigraphy (PYP/DPD) grade 2-3 + no monoclonal protein = ATTR diagnosis without biopsy
- Tafamidis (ATTR-ACT trial) reduces mortality 30%
- Avoid digoxin and CCBs (bind amyloid, toxicity)
Common Mistakes
- ❌ Assuming all amyloidosis is AL
- ❌ Using digoxin or CCBs
- ❌ Not checking free light chains/immunofixation
- ❌ Missing carpal tunnel as a clue
Last Reviewed: 2026-01-01 | MedVellum Editorial Team
Medical Disclaimer: MedVellum content is for educational purposes.